
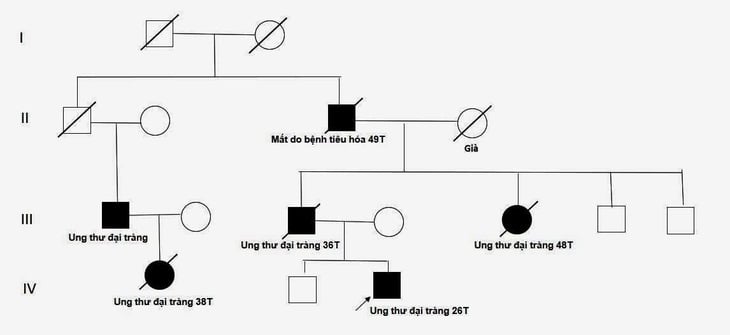
„Drzewo genealogiczne” w rodzinie 26-letniego pacjenta – zdjęcie: BVCC
Wyniki badań patologicznych wykazały, że u pacjenta występuje gruczolakorak jelita grubego.
Lekarze zwrócili większą uwagę na szczególną historię rodzinną pacjenta – jego ojciec, ciotka, wujek i kuzyn – wszyscy chorowali na raka jelita grubego i wszyscy byli hospitalizowani z powodu przedłużającego się bólu brzucha i zmęczenia.
W związku z tym pacjentowi zlecono wykonanie testu sekwencjonowania genów w celu ustalenia przyczyny. Wyniki wykazały, że pacjent jest nosicielem mutacji w genie MLH1. Był to kluczowy dowód potwierdzający rodzinne rozpoznanie zespołu Lyncha, najczęstszej postaci dziedzicznego raka jelita grubego.
Według dr Bui Bich Mai z jednostki komórek macierzystych i genów w Centrum Medycyny Nuklearnej i Onkologii (szpital Bach Mai), lekarze podczas badań spotykali się z wieloma przypadkami dziedzicznego raka, ale rodzina powyższego pacjenta to największa liczba osób z tą chorobą, z jaką lekarze kiedykolwiek się spotkali. Łącznie było 6 spokrewnionych pacjentów, w tym 5 osób z rakiem jelita grubego i 1 osoba, która zmarła w wieku 49 lat z powodu choroby przewodu pokarmowego.
„Wiele ośrodków na świecie ogłosiło metodę leczenia polegającą na wycinaniu chorych segmentów genów w celu wczesnej profilaktyki, ale metoda ta nie została jeszcze zastosowana w leczeniu raka. Jednak dzięki identyfikacji genetycznego nowotworu uwarunkowanego genami, rodzina pacjenta może zostać poddana badaniom przesiewowym w celu wcześniejszego wykrycia choroby, a leczenie będzie skuteczniejsze, na przykład poprzez wczesne badania endoskopowe i wycinanie wszystkich polipów, aby uniknąć ryzyka zachorowania na raka” – powiedział dr Mai.
Czym jest zespół Lyncha?
Zespół Lyncha to choroba genetyczna, która zwiększa ryzyko wystąpienia wielu rodzajów nowotworów, zwłaszcza raka jelita grubego. Jest spowodowana mutacjami w genach odpowiedzialnych za „naprawianie” błędów w replikacji DNA (takich jak MLH1, MSH2, MSH6, PMS2).
Gdy ten system naprawy zawodzi, mutacje szybko się kumulują, prowadząc do rozwoju raka w znacznie młodszym wieku niż normalnie. Noszenie mutacji w genie MLH1 oznacza, że dana osoba jest narażona na znacznie wyższe ryzyko rozwoju nowotworów niż populacja ogólna, w tym:
- Rak jelita grubego: ryzyko wzrasta od 1,9% do 52% - 82% (od 27 do 43 razy więcej).
- Rak endometrium (u kobiet): Ryzyko wzrasta od 1,6% do 25% - 60% (15 do 37 razy więcej).
- Rak żołądka: ryzyko wzrasta od 0,3% do 6% - 13% (20 do 43 razy więcej).
- Rak jajnika (u kobiet): Ryzyko wzrasta od 0,7% do 4% - 12% (5 do 17 razy wyższe).
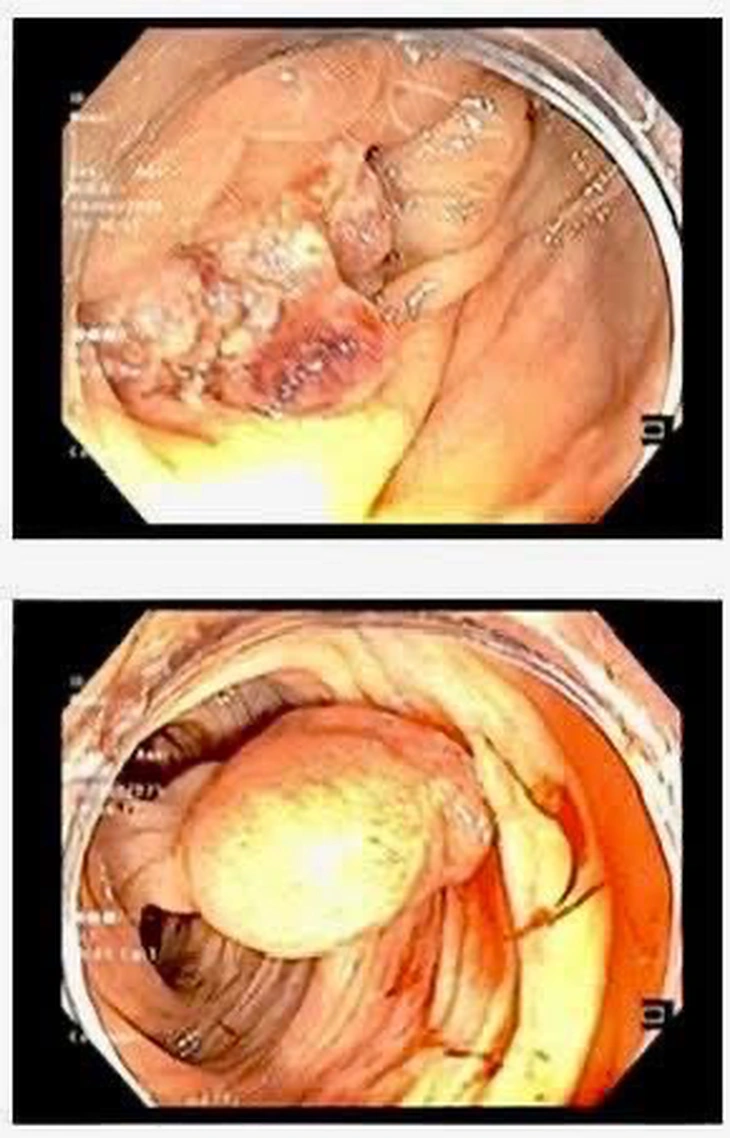
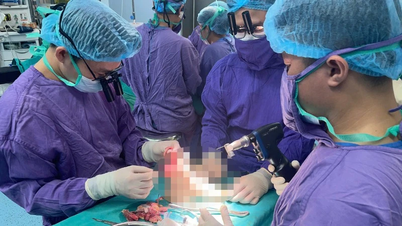
Obraz dużego guza wykrytego endoskopowo – zdjęcie: BVCC
Należy proaktywnie wcześnie wykonać badania przesiewowe w kierunku choroby, jeśli jest się nosicielem genu
Zdaniem dr Mai, dzięki zrozumieniu pierwotnej przyczyny choroby lekarze nie tylko skupiają się na leczeniu pacjentów, ale także udzielają pacjentom i ich rodzinom porad genetycznych na temat ryzyka wystąpienia chorób genetycznych.
Lekarze podkreślają również, że w przypadku rodzin, w których żyją członkowie zespołu Lyncha, niezwykle ważne jest przeprowadzenie badań genetycznych u krewnych, aby zidentyfikować nosicieli wysokiego ryzyka i dowiedzieć się, kto jest nosicielem zmutowanego genu. Takie osoby należy uważnie monitorować.
Jednocześnie należy proaktywnie badać osoby z tym genem, planując kolonoskopię co 1-2 lata od 20. do 25. roku życia oraz inne badania przesiewowe wcześniej i częściej, aby wykryć polipy lub raka we wczesnym stadium. Wczesne wykrycie i leczenie pomagają zwiększyć wskaźnik wyleczeń i poprawić jakość życia.
„U osób z rakiem występującym parami, na przykład po obu stronach jajników, lub u których w rodzinie występowało wielu krewnych z rakiem, mogą występować objawy raka dziedzicznego. Obecnie Szpital Bach Mai może precyzyjnie diagnozować raka dziedzicznego za pomocą technologii genetycznej, zapewniać poradnictwo genetyczne i leczenie osobom z tą chorobą” – dodał dr Mai.
Source: https://tuoitre.vn/vi-sao-gia-dinh-nhieu-the-he-bi-ung-thu-nguoi-tre-nhat-moi-26-tuoi-20250904214601289.htm


![[Zdjęcie] Zalewanie po prawej stronie bramy, wejście do Cytadeli Hue](https://vphoto.vietnam.vn/thumb/1200x675/vietnam/resource/IMAGE/2025/10/28/1761660788143_ndo_br_gen-h-z7165069467254-74c71c36d0cb396744b678cec80552f0-2-jpg.webp)
![[Zdjęcie] Przewodniczący Zgromadzenia Narodowego Tran Thanh Man przyjął delegację Socjaldemokratycznej Partii Niemiec](https://vphoto.vietnam.vn/thumb/1200x675/vietnam/resource/IMAGE/2025/10/28/1761652150406_ndo_br_cover-3345-jpg.webp)

![[Zdjęcie] Premier Pham Minh Chinh przewodniczył spotkaniu, na którym omawiano rozwiązania mające na celu złagodzenie skutków powodzi w prowincjach centralnych.](https://vphoto.vietnam.vn/thumb/1200x675/vietnam/resource/IMAGE/2025/10/29/1761716305524_dsc-7735-jpg.webp)





















![[Zdjęcie] Projekty dokumentów XIV Zjazdu Partii trafiają do mieszkańców Poczty Kulturalnej Komunalnej](https://vphoto.vietnam.vn/thumb/1200x675/vietnam/resource/IMAGE/2025/10/28/1761642182616_du-thao-tai-tinh-hung-yen-4070-5235-jpg.webp)































![[Infografika] Sytuacja społeczno-gospodarcza Wietnamu w ciągu 5 lat 2021-2025: Imponujące liczby](https://vphoto.vietnam.vn/thumb/402x226/vietnam/resource/IMAGE/2025/10/29/1761730747150_anh-man-hinh-2025-10-29-luc-16-38-55.png)






































Komentarz (0)