
Dies wird als Grund dafür angesehen, dass die erste Tochter des Paares, die jetzt 5 Jahre alt ist, an einer angeborenen Nebennierenhyperplasie leidet.
Laut Master, Doktor, Spezialist I Nguyen Phuong Thao, Zentrum für Fetale Medizin, Allgemeines Krankenhaus Tam Anh, Ho-Chi-Minh-Stadt, hatte das neugeborene Mädchen abnorme äußere Geschlechtsorgane, die denen eines Mannes ähnelten.
Blutuntersuchungen ergaben, dass der Spiegel von 17-Hydroxyprogesteron, einem von den Nebennieren produzierten Steroidhormon, mehr als dreimal höher als normal war, was darauf hindeutet, dass das Baby an einer Nebennierenhyperplasie litt.
Genetische Untersuchungen vor und während der Schwangerschaft spielen eine wichtige Rolle bei der Erkennung gesunder Träger von Krankheitsgenen und bei der Verhinderung der Geburt von Kindern mit schweren genetischen Erkrankungen. |
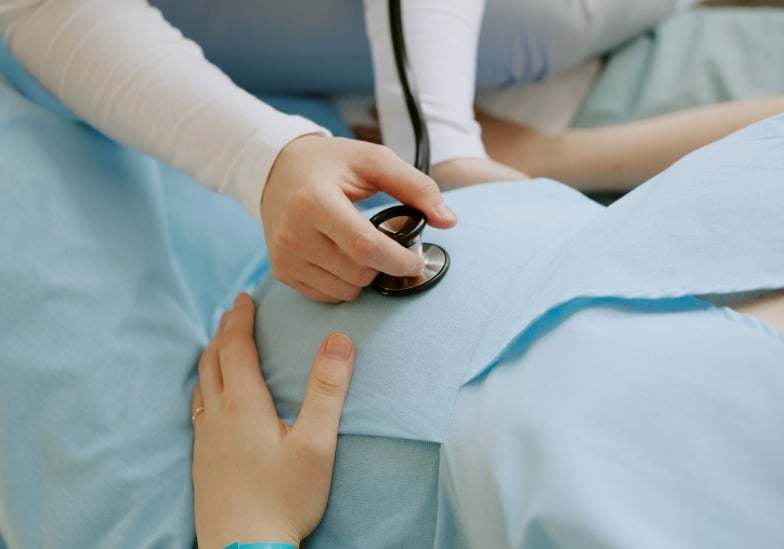
Diese Erkrankung entsteht durch eine Störung der Nebennierenrindenhormonsynthese und führt zu einer akuten Nebenniereninsuffizienz sowie zu Fehlbildungen der äußeren Geschlechtsorgane. Obwohl das Baby einen 46,XX-Chromosomensatz hat und somit weiblich ist, sind die äußeren Geschlechtsorgane vermännlicht, insbesondere die Klitoris ist vergrößert. Das Baby wurde einer plastischen Operation in Kombination mit einer endokrinen Therapie unterzogen, um diesen Zustand zu verbessern.
Die kongenitale Nebennierenhyperplasie ist eine rezessive Erbkrankheit auf Chromosom 6. Durch eine Genanalyse stellte der Arzt fest, dass Herr und Frau H. beide die CYP21A2-Genmutation tragen und symptomfreie Träger sind. Da sie vor der Schwangerschaft nicht untersucht wurden, wurde das Krankheitsgen an ihre Tochter weitergegeben.
Wird die Erkrankung frühzeitig in der Schwangerschaft erkannt, kann das Baby bereits im Mutterleib behandelt werden, wodurch eine Vermännlichung der äußeren Geschlechtsorgane verhindert werden kann. Gleichzeitig kann das Baby nach der Geburt mit Hormonen behandelt werden, um gefährliche Komplikationen wie Dehydrierung, die zum Tod führen kann, zu vermeiden.
Genetisch betrachtet hat das zweite Kind, wenn sowohl Mann als auch Frau das rezessive Gen tragen, das die Krankheit verursacht, eine 25%ige Chance, gesund geboren zu werden, ein 25%iges Risiko, an einer kongenitalen Nebennierenhyperplasie mit einem Schweregrad von mild bis schwer zu erkranken, und eine 50%ige Chance, dass das Kind ein gesunder Träger des Krankheitsgens ist, das an die nächste Generation weitergegeben werden kann.
Nachdem Ha und ihr Mann sich vor der Schwangerschaft einer genetischen Untersuchung unterzogen hatten, entschieden sie sich für eine In-vitro-Fertilisation in Kombination mit einer Embryonenuntersuchung, um die Geburt eines gesunden Babys zu gewährleisten.
Genetische Untersuchungen zur Identifizierung gesunder Träger des Krankheitsgens tragen dazu bei, die Vermännlichung der äußeren Geschlechtsorgane bei Mädchen zu verhindern oder zu minimieren. Pränatale Gentests ermöglichen Ärzten zudem eine rechtzeitige Behandlung nach der Geburt und beugen so einer akuten Nebennierenrindeninsuffizienz vor.
Werden vermännlichte Mädchen frühzeitig behandelt, können sie sich normal entwickeln; im Gegenteil, eine späte Diagnose kann zu einer vollständigen Vermännlichung führen, die später Unfruchtbarkeit und Sterilität zur Folge hat.
Jungen mit dieser Erkrankung leiden häufig unter verfrühter Pubertät und Wachstumsstörungen. Die Patienten benötigen eine lebenslange Hormonersatztherapie und die operative Korrektur von Fehlbildungen der äußeren Geschlechtsorgane.
Die kongenitale Nebennierenhyperplasie tritt bei etwa 1 von 10.000 bis 1 von 15.000 Lebendgeburten auf. Unbehandelt kann die Erkrankung schwerwiegende Symptome wie Hyperkaliämie, Erbrechen, Dehydratation, mangelnde Gewichtszunahme, niedrigen Blutdruck, Hypoglykämie, Elektrolytstörungen, Kreislaufschock und Tod zur Folge haben.
Die Krankheit führt auch zu Fehlentwicklungen der Geschlechtsorgane, wie etwa Klitorishypertrophie bei Mädchen, großem Penis oder kleinen Hoden bei Jungen sowie zu Pubertäts- und Menstruationsstörungen.
Patienten haben zudem ein erhöhtes Risiko, kardiometabolische Erkrankungen wie Bluthochdruck, Adipositas, Fettstoffwechselstörungen, Insulinresistenz und Glukosestoffwechselstörungen zu entwickeln. Werden diese Erkrankungen jedoch frühzeitig erkannt und behandelt, können die Babys ein nahezu normales Leben führen und später selbst gesunde Kinder zur Welt bringen.
Genetische Untersuchungen vor und während der Schwangerschaft spielen eine wichtige Rolle bei der Erkennung gesunder Träger von Krankheitsgenen und bei der Verhinderung der Geburt von Kindern mit schweren genetischen Erkrankungen.
Mit der Entwicklung der Fetalmedizin und der modernen Gentechnik können viele gefährliche Krankheiten bei frühzeitiger Erkennung wirksam bekämpft werden, was den Kindern und ihren Familien die Möglichkeit einer Entwicklung und eines normalen Lebens eröffnet.
Zusätzlich zu den oben genannten pathologischen Befunden ist die Thalassämie eine genetische Erkrankung, die durch eine verminderte oder unzureichende Synthese von Globinketten im Hämoglobinmolekül, einem wichtigen Bestandteil der roten Blutkörperchen, gekennzeichnet ist. Die Erkrankung umfasst zwei Hauptformen: α-Thalassämie und β-Thalassämie, je nachdem, welche Globinkette betroffen ist.
Genmutationen, die zu Globinmangel führen, können schwere Formen der Krankheit hervorrufen, die eine schwere Anämie verursachen und die Gesundheit und Entwicklung von Kindern beeinträchtigen.
Laut Dr. Luyen Thi Thanh Nga vom Medlatec Genetic Center ist der Anteil der Menschen, die das Thalassämie-Gen in Vietnam tragen, mit etwa 13,8 % aller ethnischen Gruppen recht hoch.
Insbesondere bei einigen ethnischen Minderheiten wie den Tay, Thai und Muong liegt die Prävalenz des α-Thalassämie-Gens bei über 20 %. Bei Medlatec wird im Jahr 2025 die SEA-Mutation die häufigste Form bei nachgewiesener α-Thalassämie sein.
Thalassämie ist eine rezessive Erbkrankheit. Daher zeigen Genträger oft keine äußeren Symptome, was die Diagnose ohne spezielle Tests erschwert. Sind beide Ehepartner Träger des Thalassämie-Gens, liegt das Risiko, ein Kind mit einer schweren Form der Krankheit zu bekommen, bei bis zu 25 %.
Daher ist eine genetische Untersuchung vor der Geburt oder vor der Eheschließung für die Beratung und Prävention unerlässlich. Im Fall von Frau K. und ihrem Ehemann klärte der Arzt sie gezielt über das Risiko auf, ein Kind mit schwerer Thalassämie zu bekommen, und führte eine Fruchtwasseruntersuchung zur pränatalen Diagnostik durch. Die Ergebnisse zeigten, dass der Fötus einen normalen Genotyp aufwies, was der Familie die Gewissheit gab, die Schwangerschaft fortsetzen zu können.
Ärzte empfehlen Paaren außerdem, bei nachfolgenden Schwangerschaften entweder eine natürliche Schwangerschaft in Kombination mit einer Amniozentese zur pränatalen Diagnostik oder eine In-vitro-Fertilisation in Kombination mit einem Embryonenscreening zur Risikominimierung zu wählen.
Schwere Thalassämie verursacht chronische Anämie, die lebenslange Bluttransfusionen erforderlich macht und unbehandelt leicht zu Knochenverformungen, vergrößerter Leber und Milz, Herzinsuffizienz und möglicherweise vorzeitigem Tod führen kann. Die Krankheit stellt zudem eine enorme psychische und finanzielle Belastung für Familien und die Gesellschaft dar.
Daher hilft das Thalassämie-Gen-Screening nicht nur dabei, Träger des Krankheitsgens frühzeitig zu erkennen, sondern unterstützt auch Paare bei der Familienplanung und minimiert so das Erkrankungsrisiko für zukünftige Generationen. Dies ist eine wirksame Präventionsmaßnahme und sollte in der Bevölkerung breit gestreut werden.
Quelle: https://baodautu.vn/sang-loc-truoc-sinh-giup-ngan-ngua-hieu-qua-di-tat-cho-tre-d331979.html


![[Foto] Generalsekretär To Lam empfängt den Direktor der Akademie für öffentliche Verwaltung und Volkswirtschaft beim Präsidenten der Russischen Föderation](/_next/image?url=https%3A%2F%2Fvphoto.vietnam.vn%2Fthumb%2F1200x675%2Fvietnam%2Fresource%2FIMAGE%2F2025%2F12%2F08%2F1765200203892_a1-bnd-0933-4198-jpg.webp&w=3840&q=75)










































































































Kommentar (0)